Translate this page into:
Multifocal rosai–dorfman disease
2 College of Medicine, University of Hail, Hail, Saudi Arabia
3 Department of Orthopedics, AlRazi Hospital, Kuwait Institution for Medical Specializations, Kuwait City, Kuwait, Saudi Arabia
4 Department of Pathology and Laboratory Medicine, King Abdulaziz Medical City, King Abdullah International Medical Research Center, College of Medicine, King Saud bin Abdulaziz for Health Sciences, Riyadh, Saudi Arabia
Corresponding Author:
Wazzan S Aljuhani
Department of Surgery, King Abdalaziz Medical City, King Abdalleah Research Center, King Saud Bin Abdalaziz University for Health Science, Riyadh
Saudi Arabia
aljohaniwa@ngha.med.sa
| How to cite this article: Aljuhani WS, Alobaidi SA, Alqaseer AM, Alassiri AH, Khaja AF. Multifocal rosai–dorfman disease. J Musculoskelet Surg Res 2020;4:56-60 |
Abstract
Rosai–Dorfman disease (RDD) is an uncommon proliferative histiocytic disorder whose etiology remains unknown. It is equally identified by the nomenclature sinus histiocytosis with massive lymphadenopathy. The major causes appear to be autoimmune dysregulation or proliferation of the histiocytes secondary to infection. The current diagnosis of this disorder is based on cytological and histological characteristics, with positive CD68 and S100 markers, and emperipolesis as the key diagnostic factor. We present the case of a 37-year-old man with multiple soft tissue masses in the left thigh, scalp, and back whose histological features were consistent with soft tissue RDD, which is an unusual presentation of this relatively rare disease.Introduction
Rosai–Dorfman disease (RDD) is also referred to as sinus histiocytosis with massive lymphadenopathy. RDD, known to be a disorder of benign histiocytic proliferation, has a yet-to-be unraveled etiology.[1],[2]
RDD has a deceptive onset, but its active phase appears to be extended, while its remission may be spontaneous with the possibility of a recurrence.[3]
The first description of RDD was given in 1969 by Rosai and Dorfman, identifying it as a unique clinicopathologic entity.[4] RDD is a rare disease, distributed globally with 80% of cases recorded in young adults and children.[5] RDD is slightly predominant in males (58%) and generally in people of African descent.[5] Foucar, Dorfman, and Rosai conducted the largest RDD-based study in 1990 and included 423 cases with a histopathological diagnosis of RDD.[6]
Several hypotheses have been put forward to explain the etiology of RDD. Several studies have linked this condition to infectious agents such as human herpes virus 6, Epstein–Barr virus (EBV), Nocardia, Brucella, and Klebsiella rhinoscleromatis. The major causes appear to be autoimmune dysregulation or proliferation of the histiocytes secondary to infection. The current diagnosis of this disorder is based on cytological and histological characteristics, with positive CD68 and S100 markers, and emperipolesis as the key diagnostic factor.[1],[2],[7],[8],[9],[10] Emperipolesis is a condition wherein hematopoietic cells in the living and intact state are seen in the cytoplasm of the host cells without any damage.[11] Negative CD1a markers have played an important role in differentiating them from other medical conditions.[1],[2],[7],[8],[10] Although not much is known of RDD, it has a favorable prognosis in most cases. Studies have shown that 50% of diagnosed cases usually resolve spontaneously with little or no adverse sequelae.[1],[7],[9],[10],[12] For cases requiring treatment, surgical interventions and steroids have proven effective in the treatment of severe sequelae of this disorder, including compression or obstruction of delicate organs, fever, and symptomatic lymphadenopathy.[1],[2],[8],[9].[10],[12]
The current knowledge of RDD offers a precursory framework that may facilitate the development of a standardized care model for patients. This case study includes a review of the current literature to discuss salient issues with regard to pathology, diagnosis and treatment, and prognosis of RDD.
Case Report
A 37-year-old man presented with a mass in the left thigh. It was first noticed 3 months before presentation. Until the date that he arrived at the health facility, there was no change in the size of the mass. The patient had multiple fascial scars due to a history of leishmaniasis.
Investigations
The mass measured about 10 cm × 10 cm and was located on the medial aspect of the left thigh [Figure - 1]. It was firm, mobile, and with dark discoloration of the skin. No regional lymph nodes were found to be palpable.
![[Figure - 1]](#fig_SaudiOrthopJ_2020_4_1_56_275751_f1.jpg){kind=link}
 |
| Figure 1: Computed tomography scan of the mass on the left thigh |
We did magnetic resonance imaging (MRI) of the left thigh and found a poorly defined lesion measuring 8 cm × 7 cm × 3.5 cm [Figure - 2].
![[Figure - 2]](#fig_SaudiOrthopJ_2020_4_1_56_275751_f2.jpg){kind=link}
 |
| Figure 2: Magnetic resonance imaging of the left thigh |
The location was strictly subcutaneous, being inseparable from the deep fascia invading the thigh muscles. We did not observe any deep extension. The lesion showed poorly defined margins with surrounding strands. The lesion matrix was solid, displaying intermediate signals in T1-weighted image and T2-weighted image with diffusion restriction and moderate postcontrast enhancement. MRI of the scalp demonstrated a localized subcutaneous–muscular swelling overlying the right parieto-temporal bone without locally aggressive features and without intracranial, orbital, or nasal disease manifestation. However, lumbar spine MRI showed a 1.8 x 1.8 x 1.2 cm enhancing nodule at the deep subcutaneous tissue at the level of L5-S1, which has some irregular margin, which has an appearance compatible with RDD.
Fine needle aspiration was done and showed hypocellular smears. We found that the hypocellular smears contained lymphoid cells, plasma cells, polymorphs, and blood, which was inadequate for critical cytopathological assessment.
A core needle biopsy of the thigh mass was done and showed that the mass lesion consisted of dense inflammatory infiltrate [Figure - 3], including a distinctive large and pale histiocytes with ample cytoplasm. These cells engulfed lymphocytes in a process known as emperipolesis [Figure - 4]. The background contained prominent lymphoplasmacytic cells. S100 (polyclonal) and CD163 (MRQ-26) immunostains were strongly positive in these histiocytes, and both highlight emperipolesis [Figure - 5] and [Figure - 6]; green arrows]. Immunohistochemically, large cells of the lesion were reacting negatively for CD3 (polyclonal), CD20 (L26), CD30 (Ber-H2), desmin (D33), smooth muscle actin (1A4), and CD34 (Qbend10). Kappa (polyclonal) and Lambda (polyclonal) highlight the polyclonal plasma cell component.
![[Figure - 3]](#fig_SaudiOrthopJ_2020_4_1_56_275751_f3.jpg){kind=link}
![[Figure - 4]](#fig_SaudiOrthopJ_2020_4_1_56_275751_f4.jpg){kind=link}
![[Figure - 5]](#fig_SaudiOrthopJ_2020_4_1_56_275751_f5.jpg){kind=link}
![[Figure - 6]](#fig_SaudiOrthopJ_2020_4_1_56_275751_f6.jpg){kind=link}
 |
| Figure 3: Histiocytic infiltration in a background of chronic inflammatory cells |
 |
| Figure 4: Histiocytes with abundant eosinophilic cytoplasm demonstrating emperipolesis (green arrow) |
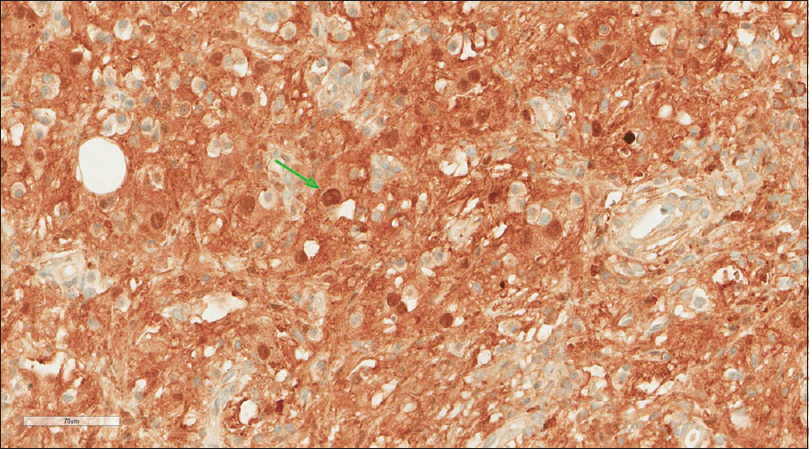 |
| Figure 5: S100 stain |
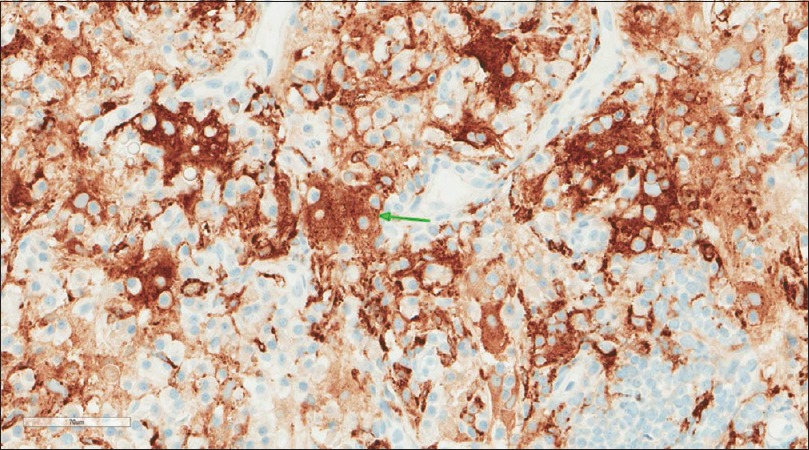 |
| Figure 6: CD163 (MRQ-26) stain |
Differential diagnosis
EBV-automated in situ hybridization was negative. Emperipolesis was highlighted nicely with S100. The final histopathological diagnosis was RDD. Positron emission tomographycomputed tomography (PETCT) scan was ordered and showed an intensely hypermetabolic left distal thigh subcutaneous mass [Figure - 7], an intensely hypermetabolic right scalp subcutaneous thickening and prominently hypermetabolic subcutaneous nodule at the S1 level that were suspicious for disease infiltration, which were consistent with RDD. Scalp and sacrolumbar spine MRIs were also ordered.
![[Figure - 7]](#fig_SaudiOrthopJ_2020_4_1_56_275751_f7.jpg){kind=link}
 |
| Figure 7: (a) An intensely hypermetabolic right scalp subcutaneous thickening and prominently subcutaneous nodule. (b) Positron emission tomography scan showing an intensely hypermetabolic left distal thigh subcutaneous mass |
Treatment
As we are a recognized oncology center that is well-equipped and is capable of dealing with bone oncology, we tackled this case. After discussing the nature of the disease with the patient, the patient was still concerned about this soft tissue mass and chose the surgical option. The patient had a wide excision of the left thigh lesion through an elliptical skin incision. The lesion was excised with the deep fascia as a safe margin. A marginal excision of the scalp lesion was done in the same setting. The patient tolerated the procedure, and the postoperative period was uneventful. The patient was discharged on the 5th postoperative day. The resected scalp mass was a white-tan soft tissue mass measuring 1.1 cm × 1.0 cm × 0.2 cm and showed aggregates of crushed lymphoid cells on histological analysis. The thigh mass confirmed the diagnosis of RDD with the same histological features described above.
Discussion
Our patient complained of a mass in the left thigh, which was first noticed 3 months before presentation. RDD is generally classified as a systemic (osseous, respiratory, or cutaneous) or nodal. In typical cases, it presents insidiously with the lymph node being infiltrated (polymorphic histiocytic), together with generalized lymphadenopathy. Cervical lymph nodes are mostly affected, together with the axillary, mediastinal, and inguinal lymph node basins.[13] RDD bears a resemblance to a malignant prognosis; however, it suffices to say that there are variations in the clinical course of this condition, ranging from spontaneous regression to progressive lymphadenopathy and extended phases of stable disease.[13] Nodular expansion is a rare complication of this disease and usually results in death. The expansion moves into vital organs and interferes with their physiology. Complications resulting in death are not detailed in existing reports.[13]
The etiology of RDD is unknown although viral agents such as EBV and herpes virus 6 are thought to contribute to the pathology of this disease via dysregulation of the immune system.[13],[14] Human herpes virus 6 was detected in situ in seven out of nine cases of sinus histiocytosis with massive lymphadenopathy by Levine et al., while Luppi et al. had demonstrated expression of human herpes virus-6 antigen by abnormal histiocytes.[15],[16] Data obtained from both studies suggest that infection by the EBV may not be causative but may contribute to the development of RDD.[17] Yoon et al. proposed that histoproliferation in RDD was an abnormal reaction of the hematolymphoid system to infection, resulting in hyperactivation of the immune system and cytosis.[18] We must emphasize that to date, the pathogenesis of RDD is still not well understood, but there are indications that it is multifactorial. Most documented cases have shown a variety of coexisting immunologically mediated disorders, such as lupus erythematosus, asthma, hemolytic anemia, and rheumatoid arthritis.[19],[20]
Systemic RDD has a higher prevalence with characteristics, including tumors at the upper respiratory tract, skin, retro-orbital tissue, and bone.[21] Adjacent soft tissue and skin are only involved in the cutaneous form of this disease without the involvement of the lymph nodes or other organs.[19] The cutaneous form of RDD occurs in a third of cases, with the neck and the head being the most affected regions.[14]
Challenges are encountered while making a differential diagnosis for this condition. Diagnosis is dependent on clinical features, radiographic features, and immunohistological analysis. Lymph nodes are usually hypermetabolic and give a positive result on PET scan. Studies show the presence of polyclonal hypergammaglobulinemia in at least 80% of patients and normochromic normocytic or hypochromic anemia in as many as 65%.[5] The histological feature of RDD includes the accumulation of histiocytes (usually proliferating histiocytes) in the lymph node sinusoids.[18] Histiocytes of RDD phagocytose lymphocytes and other cells of the immune system, resulting in emperipolesis a histological hallmark of the disease.[18] We also know that RDD is positive for CD68 and S-100 antigens while is negative for CD1a antigens.[22] It is worthy of note that the reactivity of CD1a is more common in Langerhans cell histiocytosis (LCH), while RDD features S-100 reactivity.[23] Although MRI and CT are not diagnostic of RDD, they may be vital to the assessment of local disease extension, while also excluding other possible diagnoses.[13]
The differential diagnosis of lesions' characteristic of RDD includes lymphoreticular malignancies in the presence of cervical lymphadenopathy or soft tissue sarcomas when patients present with extranodal disease. Malignant diagnosis is usually dispelled by the absence of cytologic atypia. In addition, immunohistochemistry profiles will showcase emperipolesis with macrophage-induced histiocytosis.[24] There is also the need to make a clear differentiation between LCH and RDD-similar histiocytosis.[4] Marked cytological atypia and high mitotic activity are demonstrated in malignant histiocytosis; LCH, on the other hand, appears to be CD1a positive with Birbeck granules.[4] It is also important to note that neither LCH nor malignant histiocytosis demonstrates the hallmark identification of emperipolesis.[4] Reticulohistiocytoma, although rarer, may be S100-positive. RDD, however, shows high periodic acid–Schiff-positive strain, a low number of inflammatory cells, and strong ground-glass appearance. These variations help in the provision of a definitive diagnosis.
Diagnosis of soft tissue RDD is quite challenging due to the difficulty encountered in discriminating the precise morphology of soft tissue samples. A spindled morphology is a major feature of extranodal soft tissue RDD with several collagen deposits, resulting in emperipolesis, thus making it more inconspicuous.[23] Furthermore, soft tissue RDD usually features a whorled pattern, which may cause misdiagnosis. In this case, the clinician may diagnose either malignant or benign fibrohistiocytosis.[23] Generally, cells of dermatofibrosarcoma protuberans, benign fibrous histiocytoma (all soft tissue lesions), usually have a very high nuclear ratio compared to cytoplasmic ratio, a higher number of hyperchromatic nuclei, and a unique whorled pattern compared to soft tissue RDD.[23]
Because RDD is rare and has a limited course, it has no established protocol of treatment.[13] Surgical excision is performed in symptomatic cases that are bereft of spontaneous resolution. Symptomatic cases respond successfully to steroids, interferon alpha, and alkylating agents; however, the success rates vary.[25] We still do not have a clear understanding of the role of radiotherapy. Some reports describe a full resolution while others failed to show any response whatsoever.[13]
Although there have been few descriptions of RDD in extremities, this case illustrated the importance of proper differentiation from common malignancies. It was observed that the patient tolerated the procedure, and the postoperative period was uneventful. Studies have shown that most patients with extranodal RDD usually experience a pain-free result postresection. In addition, there have been rare reports of bilateral disease development in a different site.[4],[5],[18],[26],[27],[28] Surgical excision remains the best treatment strategy for RDD lesions.[29],[30],[31]
Recommendations
There has been only one report of a local recurrence for extranodal soft tissue RDD postsurgical resection. However, there is always the possibility of a bilateral presentation, thus the need for strict clinical follow-up. There is also a need for further research to help elucidate the history of RDD and its potential for malignancy, thus facilitating the development of evidence-based therapies.
Conclusions
There is no specific agreed-upon therapy for RDD, owing to the rarity of the condition and the difficulty in preoperative diagnosis. Surgical resection proves to be the most successful strategy in preventing a recurrence of the condition. Because of the indolence and rarity of the condition, we cannot endorse surveillance; however, it is important to consider extranodal soft tissue RDD as part of the differential diagnosis of patients presenting with masses of soft tissue whose origin seems suspicious.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that his name and initials will not be published, and due efforts will be made to conceal his identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Authors' contributions
AFK and SAA collected the necessary data and conducted the data cleansing process. WSA is the primary surgeon and the consultant in charge of the patient care. Also, WSA built the team for starting and initiating the case report. AHA diagnosed the case and provided the pathology description and images in the manuscript. AMA and SAA were responsible for patient counseling regarding the consent. AFK produced the initial and final manuscript. All authors have critically reviewed and approved the final draft and are responsible for the content and similarity index of the manuscript.
| 1. | Lima FB, Barcelos PS, Constâncio AP, Nogueira CD, Melo-Filho AA. Rosai-Dorfman disease with spontaneous resolution: Case report of a child. Rev Bras Hematol Hemoter 2011;33:312-4. [Google Scholar] |
| 2. | Agarwal A, Pathak S, Gujral S. Sinus histiocytosis with massive lymphadenopathy– A review of seven cases. Indian J Pathol Microbiol 2006;49:509-15. [Google Scholar] |
| 3. | AlWadani S, Robinson S, Myers R, Akpek EK, Eberhart CG. No increase in IgG4-positive plasma cells in limbal Rosai-Dorfman disease. Cornea 2014;33:844-7. [Google Scholar] |
| 4. | Kong YY, Kong JC, Shi DR, Lu HF, Zhu XZ, Wang J, et al. Cutaneous Rosai-Dorfman disease: A clinical and histopathologic study of 25 cases in China. Am J Surg Pathol 2007;31:341-50. [Google Scholar] |
| 5. | Sodhi KS, Suri S, Nijhawan R, Kang M, Gautam V. Rosai-Dorfman disease: Unusual cause of diffuse and massive retroperitoneal lymphadenopathy. Br J Radiol 2005;78:845-7. [Google Scholar] |
| 6. | Penna Costa AL, Oliveira e Silva N, Motta MP, Athanazio RA, Athanazio DA, Athanazio PRF. Soft tissue Rosai–Dorfman disease of the posterior mediastinum. J Bras Pneumol 2009;35:717-20. [Google Scholar] |
| 7. | Pradhananga RB, Dangol K, Shrestha A, Baskota DK. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): A case report and literature review. Int Arch Otorhinolaryngol 2014;18:406-8. [Google Scholar] |
| 8. | Ha H, Kim KH, Ahn YJ, Kim JH, Kim JE, Yoon SS. A rare case of Rosai-Dorfman disease without lymphadenopathy. Korean J Intern Med 2016;31:802-4. [Google Scholar] |
| 9. | McClain KL, Natkunam Y, Swerdlow SH. Atypical cellular disorders. Hematology 2004;2004:283-96. [Google Scholar] |
| 10. | Dalia S, Sagatys E, Sokol L, Kubal T. Rosai-Dorfman disease: Tumor biology, clinical features, pathology, and treatment. Cancer Control 2014;21:322-7. [Google Scholar] |
| 11. | Amita K, Vijay Shankar S, Abhishekh MG, Geethalakshmi U. Emperipolesis in a case of adult T cell lymphoblastic lymphoma (mediastinal type) – Detected at FNAC and imprint cytology. Online J Health Allied Sci 2011;10:11. [Google Scholar] |
| 12. | Sachdev R, Setia N, Jain S. Sinus histiocytosis with massive lymphadenopathy. Is the lymph node enlargement always massive?. Med Oral Patol Oral Cir Bucal 2007;12:E198-200. [Google Scholar] |
| 13. | Pinto DCG, Vidigal TA, Castro B, Santos BH, DeSousa NJA. Rosai–Dorfman disease in the differential diagnosis of cervical lymphadenopathy. Bras J Otorrinolaringol 2008;74:632-5. [Google Scholar] |
| 14. | Ensari S, Selcuk A, Dere H, Perez N, Dizbay Sak S. Rosai–Dorfman disease presenting as laryngeal masses. Kulak Burun Bogaz Ihtis Derg 2008;18:110-4. [Google Scholar] |
| 15. | Levine PH, Jahan N, Murari P, Manak M, Jaffe ES. Detection of human herpesvirus 6 in tissues involved by sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease). J Infect Dis 1992;166:291-5. [Google Scholar] |
| 16. | Luppi M, Barozzi P, Garber R, Maiorana A, Bonacorsi G, Artusi T, et al. Expression of human herpesvirus-6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol 1998;153:815-23. [Google Scholar] |
| 17. | Govender D, Chetty R. Inflammatory pseudotumour and Rosai-Dorfman disease of soft tissue: a histological continuum? J Clin Pathol 1997;50:79-81. [Google Scholar] |
| 18. | Yoon AJ, Parisien M, Feldman F, Young-In Lee F. Extranodal Rosai-Dorfman disease of bone, subcutaneous tissue and paranasal sinus mucosa with a review of its pathogenesis. Skeletal Radiol 2005;34:653-7. [Google Scholar] |
| 19. | Potts CA, Bozeman AP, Walker AN, Floyd WE 3rd. Cutaneous Rosai-Dorfman disease of the forearm: Case report. J Hand Surg Am 2008;33:1409-13. [Google Scholar] |
| 20. | Stebbing C, van der Walt J, Ramadan G, Inusa B. Rosai-Dorfman disease: A previously unreported association with sickle cell disease. BMC Clin Pathol 2007;7:3. [Google Scholar] |
| 21. | Woodcock RJ Jr., Mandell JW, Lipper MH. Sinus histiocytosis (Rosai-Dorfman disease) of the suprasellar region: MR imaging findings – A case report. Radiology 1999;213:808-10. [Google Scholar] |
| 22. | Lu D, Estalilla OC, Manning JT Jr., Medeiros LJ. Sinus histiocytosis with massive lymphadenopathy and malignant lymphoma involving the same lymph node: A report of four cases and review of the literature. Mod Pathol 2000;13:414-9. [Google Scholar] |
| 23. | Montgomery EA, Meis JM, Frizzera G. Rosai-Dorfman disease of soft tissue. Am J Surg Pathol 1992;16:122-9. [Google Scholar] |
| 24. | Konishi E, Ibayashi N, Yamamoto S, Scheithauer BW. Isolated intracranial Rosai-Dorfman disease (sinus histiocytosis with massive lymphadenopathy). AJNR Am J Neuroradiol 2003;24:515-8. [Google Scholar] |
| 25. | Podberezin M, Angeles R, Guzman G, Peace D, Gaitonde S. Primary pancreatic sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): An unusual extranodal manifestation clinically simulating malignancy. Arch Pathol Lab Med 2010;134:276-8. [Google Scholar] |
| 26. | Molina-Garrido MJ, Guillén-Ponce C. Extranodal Rosai-Dorfman disease with cutaneous and periodontal involvement: A rare presentation. Case Rep Oncol 2011;4:96-100. [Google Scholar] |
| 27. | Komaragiri M, Sparber LS, Santos-Zabala ML, Dardik M, Chamberlain RS. Extranodal Rosai-Dorfman disease: A rare soft tissue neoplasm masquerading as a sarcoma. World J Surg Oncol 2013;11:63. [Google Scholar] |
| 28. | Suster S, Cartagena N, Cabello-Inchausti B, Robinson MJ. Histiocytic lymphophagocytic panniculitis. An unusual extranodal presentation of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Arch Dermatol 1988;124:1246-9. [Google Scholar] |
| 29. | Kemson R, Rouse R. Surgical pathology criteria. Inflammatory Myofibroblastic Tumor. Stanford, CA: Stanford School of Medicine; 2008. [Google Scholar] |
| 30. | Sabel MS. From sarcomas of bone and soft tissue. In: Greenfield LJ, Mulholland MW, editors. Greenfield's Surgery: Scientific Principles and Practice. Vol. 5. Philadelphia, PA: Wolters Kluwer Health/Lippincott William and Wilkins; 2011. p. 2151-76. [Google Scholar] |
| 31. | Shi XY, Ma DL, Fang K. Cutaneous Rosai-Dorfman disease presenting as a granulomatous rosacea-like rashs. Chin Med J (Engl) 2011;124:793-4. [Google Scholar] |
Fulltext Views
1,658
PDF downloads
293




